Assembling TSS annotations#
MIRA allows users to flexibly re-define the TSS locations of genes for RP modeling. Whether you want to compile annotations for a different sequence version or a new organism, you can follow these instructions to produce a non-redundant annotation of genes’ canonical transcription start sites.
First, go the UCSC table browser, and fill in the dataset selection form. Blue indicates where you must change the fields for your annotation, Red shows which fields must be set.
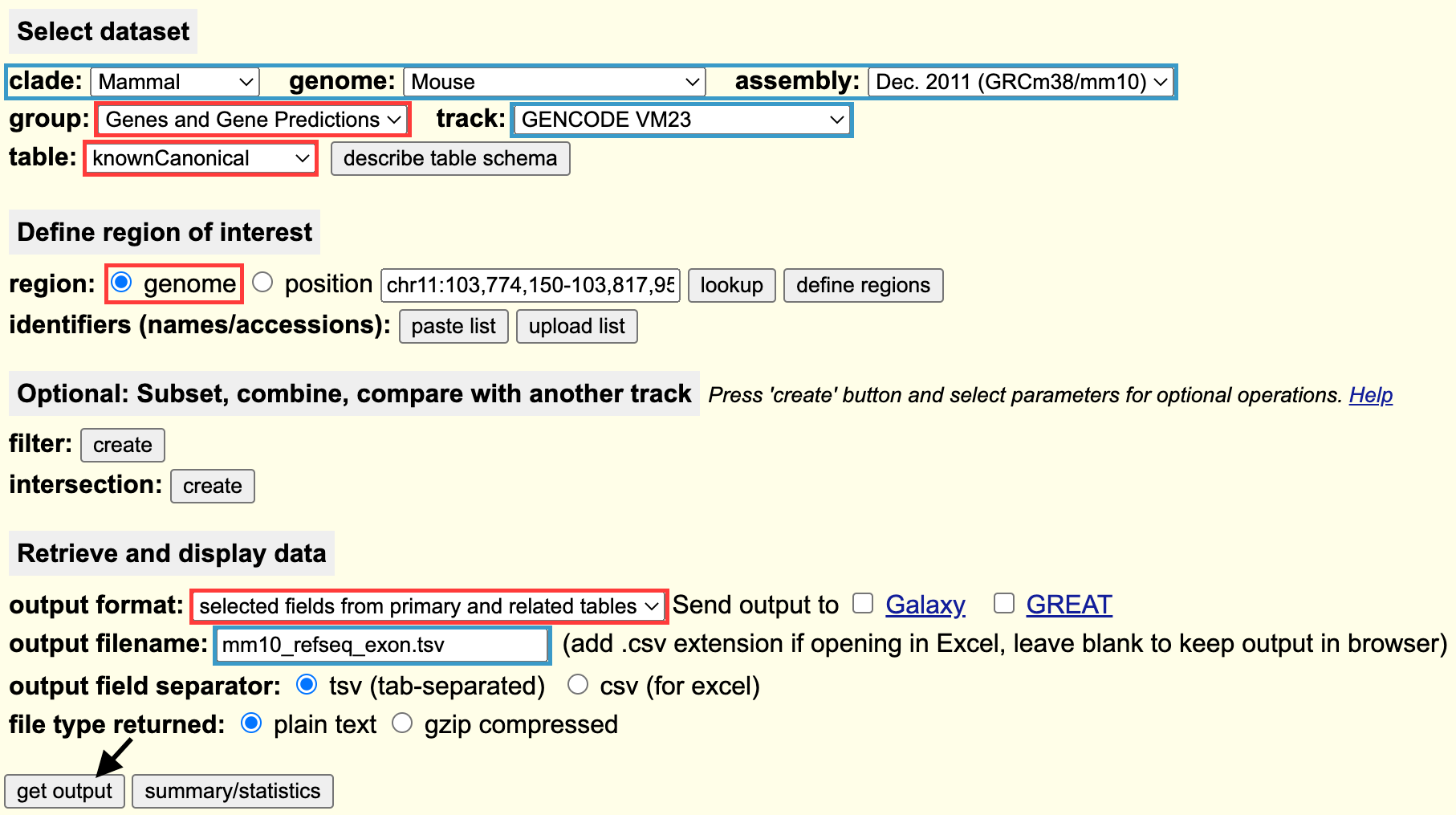
The “knownCanonical” table contains the canonical TSS and splice variants for each gene. Click get output to continue.
On the next page, scroll down to the Linked Tables box, and check the kgXref and knownGene tables. Click allow selection from checked tables. Scroll back up to the top.
Now, check the following fields to join the gene symbol and exon data to the knownCanonical table:

Again, hit get output (in the top box), and this will download the annotation. Now, there is a little bit of data cleaning to do.
First, load the data that was just downloaded:
[1]:
import pandas as pd
[2]:
tss_data = pd.read_csv('~/Downloads/mm10_refseq_exon (1).tsv', sep = '\t')
This data contains the canonical splice variants for each gene, and contains mostly unique records in terms of gene symbols.
Now clean up the column names, and drop the remaining duplicates on the geneSymbol field:
[3]:
tss_data.columns = tss_data.columns.str.split('.').str.get(-1)
tss_data['geneSymbol'] = tss_data.geneSymbol.str.upper()
tss_data = tss_data.drop_duplicates(subset='geneSymbol')
This data is most useful for other programs (pygenometracks, bedtools, etc.) in bed12 format, so we have to do a little munging. The code below converts the absolute coordinates of the exonStarts and exonEnds columns to relative coordinates based on the TSS of the gene:
[4]:
def get_block_sizes(row):
if row.exonCount <= 1:
return str(int(row.exonEnds) - int(row.exonStarts))
else:
return ','.join(map(
lambda x : str(int(x[1]) - int(x[0])),
zip(row['exonStarts'].split(','), row['exonEnds'].split(','))
))
def relative_start_pos(row):
starts = map(str, row['exonStarts'].split(','))
return ','.join(map(
lambda exon_start : str(int(exon_start) - int(row['txStart'])),
starts
))
tss_data['exonEnds'] = tss_data.exonEnds.str.strip(',')
tss_data['exonStarts'] = tss_data.exonStarts.str.strip(',')
tss_data['blockSizes'] = tss_data.apply(get_block_sizes, axis = 1)
tss_data['blockStarts'] = tss_data.apply(relative_start_pos,
axis = 1)
Add some fake columns to make a valid bed12 file:
[5]:
tss_data['score'] = 0
tss_data['thickStart'] = 0
tss_data['thickEnd'] = 0
tss_data['itemRGB'] = 0
And finally, write to disk as a tsv:
[6]:
relevant_cols = ['chrom','txStart','txEnd','geneSymbol','score','strand',
'thickStart','thickEnd','itemRGB','exonCount','blockSizes','blockStarts'] # bed12 column order
tss_data = tss_data.sort_values(['chrom','txStart']) # sort by chrom, start position \
tss_data = tss_data.reset_index()[relevant_cols] # extract ordered columns
tss_data.to_csv('data/tss_data.bed', # save name
header=['#' + x for x in relevant_cols], # add a "#" before each column name so they are ignored by other programs
index=None, # don't write the index
sep = '\t') # make it a .tsv!
[1]:
!head -n3 data/tss_data.bed
#chrom #txStart #txEnd #geneSymbol #score #strand #thickStart #thickEnd #itemRGB #exonCount #blockSizes #blockStarts
chr1 3073252 3074322 4933401J01RIK 0 + 0 0 0 1 1070 0
chr1 3102015 3102125 GM26206 0 + 0 0 0 1 110 0
Depending on your data source, you may need to change some column names, but following these steps will yield a non-redundant dataset of gene TSS and exon locations that are useful for MIRA and many other programs.