mira.topics.ExpressionTopicModel#
- class mira.topics.ExpressionTopicModel(*args, **kwargs)#
Generic class for topics models for analyzing gene expression data. All GEX topic models inherit from this class and implement the same methods.
- Attributes
- featuresnp.ndarray[str]
Array of exogenous feature names, all features used in learning topics
- highly_variablenp.ndarray[boolean]
Boolean array marking which features were “highly_variable”/endogenous, used to train encoder
- encodertorch.nn.Sequential
Encoder neural network
- decodertorch.nn.Sequential
Decoder neural network
- num_exog_featuresint
Number of exogenous features to predict using decoder network
- num_endog_featuresint
Number of endogenous feature used for encoder network
- devicetorch.device
Device on which model is allocated
- enrichmentsdict
Results from enrichment analysis of topics. For expression topic model, this gives geneset enrichments from Enrichr. For accessibility topic model, this gives motif enrichments.
topic_colslistAttribute, returns the names of the columns for the topics added by the predict method to an anndata object.
Methods
fetch_enrichments([ontologies])Iterate through all topics and fetch enrichments.
fetch_topic_enrichments(topic_num[, ontologies])Fetch Enrichr enrichments for a topic.
fit(adata_or_dirname[, writer, reinit, ...])Initializes new weights, then fits model to data.
get_enrichments(topic_num)Return the enrichment results for a given topic.
get_hierarchical_umap_features(adata[, key, ...])Leverage the hiearchical relationship between topic-feature distributions to prduce a balance matrix for isometric logratio transformation.
get_learning_rate_bounds(adata_or_dirname[, ...])Use the learning rate range test (LRRT) to determine minimum and maximum learning rates that enable the model to traverse the gradient of the loss.
get_top_genes(topic_num[, top_n, min_genes, ...])For a module, return the top n genes that are most activated.
get_umap_features(adata[, key, box_cox, add_key])Predict transformed topic compositions for each cell to derive nearest- neighbors graph.
impute(adata[, key, batch_size, bar, ...])Impute the relative frequencies of features given the cells' topic compositions.
instantiate_model(adata_or_dirname)Given the exogenous and enxogenous features provided, instantiate weights of neural network.
load(filename)Load a pre-trained topic model from disk.
plot_enrichments(topic_num[, show_genes, ...])Make plot of geneset enrichments results.
plot_learning_rate_bounds([figsize, ax, ...])Plot the loss vs.
post_topic(topic_num[, top_n, min_genes, ...])Post the top genes from topic to Enrichr for geneset enrichment analysis.
post_topics([top_n, min_genes, max_genes])Iterate through all topics and post top genes to Enrichr.
predict(adata_or_dirname[, batch_size, bar, ...])Predict the topic compositions of cells in the data.
rank_genes(topic_num)Ranks genes according to their activation in module topic_num.
rank_modules(gene)For a gene, rank how much its expression is activated by each module
save(filename)Save topic model.
score(adata_or_dirname[, batch_size])Get normalized ELBO loss for data.
set_device(device)Move topic model to a new device.
set_learning_rates(min_lr, max_lr)Set the lower and upper learning rate bounds for the One-cycle learning rate policy.
to_cpu()Move topic model to CPU device "cpu", if available.
to_gpu()Move topic model to GPU device "cuda:0", if available.
trim_learning_rate_bounds([...])Adjust the learning rate boundaries for the One-cycle learning rate policy.
- get_learning_rate_bounds(adata_or_dirname, num_steps=100, eval_every=3, num_epochs=3, lower_bound_lr=1e-06, upper_bound_lr=5)#
Use the learning rate range test (LRRT) to determine minimum and maximum learning rates that enable the model to traverse the gradient of the loss.
Steps through linear increase in log-learning rate from lower_bound_lr to upper_bound_lr while recording loss of model. Learning rates which produce greater decreases in model loss mark range of possible learning rates.
- Parameters
- adataanndata.AnnData
AnnData of expression or accessibility features to model
- num_stepsint, default=100
Number of steps to run the LRRT.
- eval_everyint, default=3,
Aggregate this number of batches per evaluation of the objective loss. Larger numbers give lower variance estimates of model performance.
- lower_bound_lrfloat, default=1e-6
Start learning rate of LRRT
- upper_bound_lrfloat, default=5
End learning rate of LRRT
- Returns
- min_learning_ratefloat
Lower bound of estimated range of optimal learning rates
- max_learning_ratefloat
Upper bound of estimated range of optimal learning rates
Examples
>>> model.get_learning_rate_bounds(rna_data, num_epochs = 3) Learning rate range test: 100%|██████████| 85/85 [00:17<00:00, 4.73it/s] (4.619921114045972e-06, 0.1800121741235493)
- set_learning_rates(min_lr, max_lr)#
Set the lower and upper learning rate bounds for the One-cycle learning rate policy.
- Parameters
- min_lrfloat
Lower learning rate boundary
- max_lrfloat
Upper learning rate boundary
- Returns
- None
- trim_learning_rate_bounds(lower_bound_trim=0.0, upper_bound_trim=0.5)#
Adjust the learning rate boundaries for the One-cycle learning rate policy. The lower and upper bounds should span the learning rates with the greatest downwards slope in loss.
- Parameters
- lower_bound_trimfloat>=0, default=0.
Log increase in learning rate of lower bound relative to estimated boundary given from LRRT. For example, if the estimated boundary by LRRT is 1e-4 and user gives `lower_bound_trim`=1, the new lower learning rate bound is set at 1e-3.
- upper_bound_trimfloat>=0, default=0.5,
Log decrease in learning rate of upper bound relative to estimated boundary give from LRRT.
- Returns
- min_learning_ratefloat
Lower bound of estimated range of optimal learning rates
- max_learning_ratefloat
Upper bound of estimated range of optimal learning rates
Examples
>>> model.trim_learning_rate_bounds(2, 1) (4.619921114045972e-04, 0.1800121741235493e-1)
- plot_learning_rate_bounds(figsize=(10, 7), ax=None, show_spline=True)#
Plot the loss vs. learning rate curve generated by the LRRT test with the current boundaries.
- Parameters
- figsizetuple(int, int), default=(10,7)
Size of the figure
- axmatplotlib.pyplot.axes or None, default = None
Pre-supplied axes for plot. If None, will generate new axes
- Returns
- axmatplotlib.pyplot.axes
Examples
When setting the learning rate bounds for the topic model, first run the LLRT test with get_learning_rate_bounds. Then, set the bounds on their respective ends of the part of the LRRT plot with the greatest slope, like below.
>>> model.trim_learning_rate_bounds(5,1) # adjust the bounds >>> model.plot_learning_rate_bounds()

If the LRRT line appears to vary cyclically, that means your batches may be not be independent of each other (perhaps batches later in the epoch are more similar to eachother and more difficult than earlier batches. If this happens, randomize the order of your input data using.
- instantiate_model(adata_or_dirname)#
Given the exogenous and enxogenous features provided, instantiate weights of neural network. Called internally by fit.
- Parameters
- adataanndata.AnnData
AnnData of expression or accessibility features to model
- Returns
- selfobject
- fit(adata_or_dirname, writer=None, reinit=True, log_every=10)#
Initializes new weights, then fits model to data.
- Parameters
- adataanndata.AnnData
AnnData of expression or accessibility features to model
- writertorch.utils.tensorboard.SummarWriter object or mira.topics.Writer object
Tracks per-batch loss metrics during training. The tensorboard object writes tensorboard log files, which can be visualized using TensorBoard. The MIRA Writer just tracks values in an in-memory dictionary.
- log_everyint, default = 10
Average the batch metrics every “log_every” steps.
- reinitbooleandefault = True
If fitting an already-trained model, whether to re-initialize weights with random values or keep the existing weights.
- Returns
- selfobject
Fitted topic model
Note
Fitting a topic model usually takes a 2-5 minutes for the expression model and 5-10 minutes for the accessibility model for a typical experiment (5K to 40K cells).
Tuning the topic model hyperparameters, however, can take longer. Finding the best number of topics significantly increases the interpretability of the model and its faithfullness to the underlying biology and is well worth the wait. To learn about topic model tuning, see mira.topics.TopicModelTuner.
- predict(adata_or_dirname, batch_size=256, bar=True, box_cox=0.25, add_key='X_topic_compositions', add_cols=True, col_prefix='topic_')#
Predict the topic compositions of cells in the data. Adds the topic compositions to the .obsm field of the adata object.
- Parameters
- adataanndata.AnnData
AnnData of expression or accessibility features to model
- batch_sizeint>0, default=512
Minibatch size to run cells through encoder network to predict topic compositions. Set to highest value where tensors will fit in memory to increase speed.
- Returns
- adataanndata.AnnData
- .obsm[‘X_topic_compositions’]np.ndarray[float] of shape (n_cells, n_topics)
Topic compositions of cells
- .obsm[‘X_umap_features’]np.ndarray[float] of shape (n_cells, n_topics)
ILR transformed topic embeddings for use with nearest neighbors graph
- .obs[‘topic_1’] … .obs[‘topic_N’]np.ndarray[float] of shape (n_cells,)
Columns for the activation of each topic.
Examples
>>> model.predict(adata) Predicting latent vars: 100%|█████████████████████████| 36/36 [00:03<00:00, 9.29it/s] INFO:mira.adata_interface.topic_model:Added key to obsm: X_topic_compositions INFO:mira.adata_interface.topic_model:Added cols: topic_1, topic_2, topic_3, topic_4, topic_5 >>> scanpy.pp.neighbors(adata, metric = "manhattan", use_rep = "X_umap_features") >>> scanpy.tl.umap(adata, min_dist 0.1) >>> scanpy.pl.umap(adata, color = model.topic_cols, **mira.pref.topic_umap(ncols = 3))
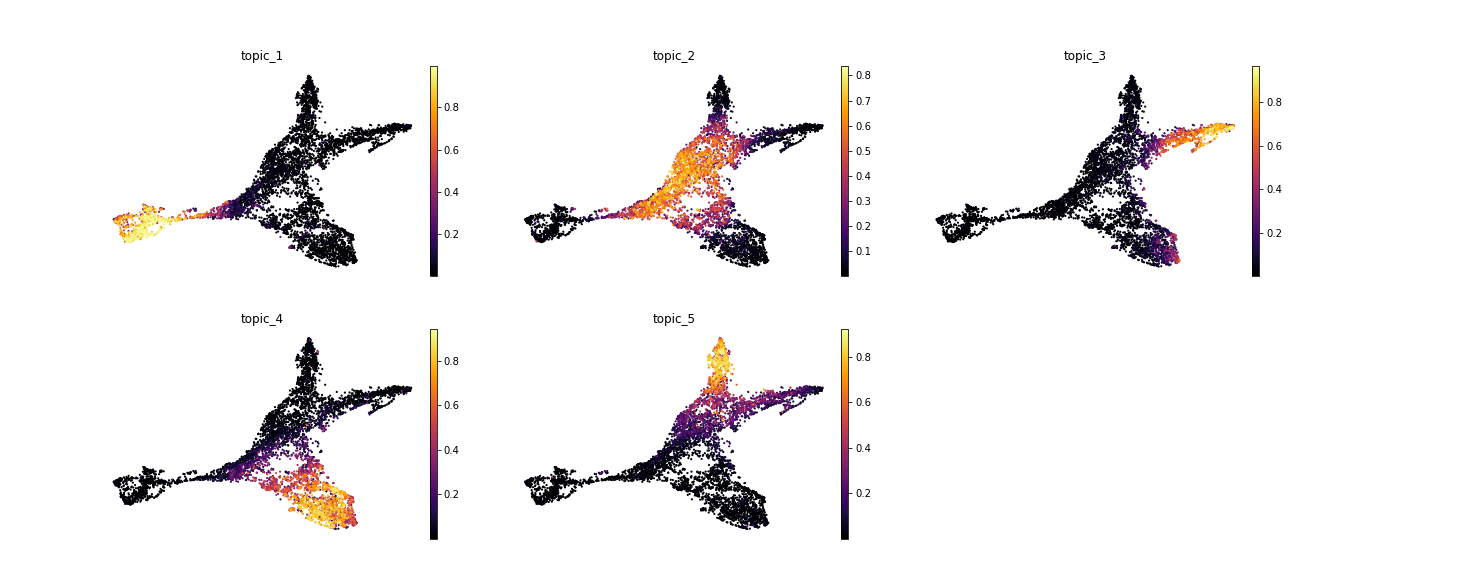
- get_hierarchical_umap_features(adata, key='X_topic_compositions', dendogram_key='topic_dendogram', box_cox=0.5, *, add_key='X_umap_features')#
Leverage the hiearchical relationship between topic-feature distributions to prduce a balance matrix for isometric logratio transformation. The function get_umap_features uses an arbitrary balance matrix that does not account for the relationship between topics.
The “UMAP features” are the transformation of the topic compositions that encode biological similarity in the high-dimensional space. Use those features to produce a joint KNN graph for downstream analysis using UMAP, clustering, and pseudotime inference.
- Parameters
- adataanndata.AnnData
AnnData of expression or accessibility features to model
- box_cox“log” or float between 0 and 1
Constant for box-cox transformation of topic compositions
- Returns
- adataanndata.AnnData
- .obsm[‘X_umap_features’]np.ndarray[float] of shape (n_cells, n_topics)
Transformed topic compositions of cells
- .uns[‘topic_dendogram’]np.ndarray
linkage matrix given by scipy.cluster.hierarchy.linkage of hiearchical relationship between topic-feature distributions. Hierarchy calculated by hellinger distance and ward linkage.
Examples
>>> model.get_hierarchical_umap_features(adata) # to make features INFO:mira.adata_interface.topic_model:Fetching key X_topic_compositions from obsm INFO:mira.adata_interface.core:Added key to obsm: X_umap_features INFO:mira.adata_interface.topic_model:Added key to uns: topic_dendogram >>> scanpy.pp.neighbors(adata, metric = "manhattan", use_rep = "X_umap_features") # to make KNN graph
- get_umap_features(adata, key='X_topic_compositions', box_cox=0.5, *, add_key='X_umap_features')#
Predict transformed topic compositions for each cell to derive nearest- neighbors graph. Projects topic compositions to orthonormal space using isometric logratio transformation.
The “UMAP features” are the transformation of the topic compositions that encode biological similarity in the high-dimensional space. Use those features to produce a joint KNN graph for downstream analysis using UMAP, clustering, and pseudotime inference.
- Parameters
- adataanndata.AnnData
AnnData of expression or accessibility features to model
- box_cox“log” or float between 0 and 1
Constant for box-cox transformation of topic compositions
- Returns
- adataanndata.AnnData
- .obsm[‘X_umap_features’]np.ndarray[float] of shape (n_cells, n_topics)
Transformed topic compositions of cells
Examples
>>> model.get_umap_features(adata) # to make features INFO:mira.adata_interface.topic_model:Fetching key X_topic_compositions from obsm INFO:mira.adata_interface.core:Added key to obsm: X_umap_features >>> scanpy.pp.neighbors(adata, metric = "manhattan", use_rep = "X_umap_features") # to make KNN graph
- score(adata_or_dirname, batch_size=256)#
Get normalized ELBO loss for data. This method is only available on topic models that have not been loaded from disk.
- Parameters
- adataanndata.AnnData
AnnData of expression or accessibility features to model
- batch_sizeint>0, default=512
Minibatch size to run cells through encoder network to predict topic compositions. Set to highest value where tensors will fit in memory to increase speed.
- Returns
- lossfloat
- Raises
- AttributeError
If attempting to run after loading from disk or before training.
Notes
The score method is only available after training a topic model. After saving and writing to disk, this function will no longer work.
Examples
>>> model.score(rna_data) 0.11564
- impute(adata, key='X_topic_compositions', batch_size=256, bar=True, add_layer='imputed', sparse=False)#
Impute the relative frequencies of features given the cells’ topic compositions. The value given is rho (see manscript for details).
- Parameters
- adataanndata.AnnData
AnnData of expression or accessibility features to model
- batch_sizeint>0, default=512
Minibatch size to run cells through encoder network to predict topic compositions. Set to highest value where tensors will fit in memory to increase speed.
- Returns
- anndata.AnnData
- .layers[‘imputed’]np.ndarray[float] of shape (n_cells, n_features)
Imputed relative frequencies of features
Examples
>>> model.impute(rna_data) >>> rna_data View of AnnData object with n_obs × n_vars = 18482 × 22293 layers: 'imputed'
- save(filename)#
Save topic model.
- Parameters
- filenamestr
File name to save topic model, recommend .pth extension
- to_gpu()#
Move topic model to GPU device “cuda:0”, if available.
- to_cpu()#
Move topic model to CPU device “cpu”, if available.
- set_device(device)#
Move topic model to a new device.
- Parameters
- devicestr
Name of device on which to allocate model
- classmethod load(filename)#
Load a pre-trained topic model from disk.
- Parameters
- filenamestr
File name of saved topic model
Examples
>>> rna_model = mira.topics.ExpressionTopicModel.load('rna_model.pth') >>> atac_model = mira.topics.AccessibilityTopicModel.load('atac_model.pth')
- rank_genes(topic_num)#
Ranks genes according to their activation in module topic_num. Sorted from least to most activated.
- Parameters
- topic_numint
For which module to rank genes
- Returns
- np.ndarray: sorted array of gene names in order from most suppressed to most activated given the specified module
Examples
Genes are ranked from least to most activated. To get the top genes:
>>> rna_model.rank_genes(0)[-10:] array(['ESRRG', 'APIP', 'RPGRIP1L', 'TM4SF4', 'DSCAM', 'NRAD1', 'ST3GAL1', 'LEPR', 'EXOC6', 'SLC44A5'], dtype=object)
- rank_modules(gene)#
For a gene, rank how much its expression is activated by each module
- Parameters
- genestr
Name of gene
- Returns
- listof format [(topic_num, activation), …]
- Raises
- AssertionError: if gene is not in self.genes
Examples
To see the top 5 modules associated with gene “GHRL”:
>>> rna_model.rank_modules('GHRL')[:5] [(14, 3.375548), (22, 2.321417), (1, 2.3068447), (0, 1.780294), (9, 1.3936363)]
- get_top_genes(topic_num, top_n=None, min_genes=200, max_genes=600)#
For a module, return the top n genes that are most activated.
- Parameters
- topic_numint
For which module to return most activated genes
- top_nint > 0
Number of genes to return
- min_genesint > 0
If top_n is None, all activations (distributed standard normal) greater than 3 will be posted. If this is less than min_genes, then min_genes will be posted.
- max_genesint > 0
If top_n is None, a maximum of max_genes will be posted.
- Returns
- np.ndarrayNames of top n genes, sorted from least to most activated
- post_topic(topic_num, top_n=500, min_genes=200, max_genes=600)#
Post the top genes from topic to Enrichr for geneset enrichment analysis.
- Parameters
- topic_numint
Topic number to post geneset
- top_nint, default = 500
Number of genes to post
- min_genesint > 0
If top_n is None, all activations (distributed standard normal) greater than 3 will be posted. If this is less than min_genes, then min_genes will be posted.
- max_genesint > 0
If top_n is None, a maximum of max_genes will be posted.
Examples
>>> rna_model.post_topic(10, top_n = 500) >>> rna_model.post_topic(10)
- post_topics(top_n=500, min_genes=200, max_genes=600)#
Iterate through all topics and post top genes to Enrichr.
- Parameters
- top_nint, default = 500
Number of genes to post
- min_genesint > 0
If top_n is None, all activations (distributed standard normal) greater than 3 will be posted. If this is less than min_genes, then min_genes will be posted.
- max_genesint > 0
If top_n is None, a maximum of max_genes will be posted.
Examples
>>> rna_model.post_topics()
- fetch_topic_enrichments(topic_num, ontologies=['WikiPathways_2019_Human', 'WikiPathways_2019_Mouse', 'KEGG_2019_Human', 'KEGG_2019_Mouse', 'GO_Molecular_Function_2018', 'GO_Cellular_Component_2018', 'GO_Biological_Process_2018', 'BioPlanet_2019'])#
Fetch Enrichr enrichments for a topic. Will return results for the ontologies listed.
- Parameters
- topic_numint
Topic number to fetch enrichments
- ontologieslist[str], default = mira.tools.enrichr_enrichments.LEGACY_ONTOLOGIES
List of ontology names from which to retrieve results. May provide a list of any onlogies hosted on Enrichr.
Examples
>>> rna_model.fetch_topic_enrichments(10, ontologies = ['WikiPathways_2019_Mouse'])
- fetch_enrichments(ontologies=['WikiPathways_2019_Human', 'WikiPathways_2019_Mouse', 'KEGG_2019_Human', 'KEGG_2019_Mouse', 'GO_Molecular_Function_2018', 'GO_Cellular_Component_2018', 'GO_Biological_Process_2018', 'BioPlanet_2019'])#
Iterate through all topics and fetch enrichments.
- Parameters
- ontologieslist[str], default = mira.tl.LEGACY_ONTOLOGIES
List of ontology names from which to retrieve results. May provide a list of any onlogies hosted on Enrichr.
Examples
>>> rna_model.fetch_enrichments(ontologies = ['WikiPathways_2019_Mouse'])
- get_enrichments(topic_num)#
Return the enrichment results for a given topic.
- Returns
- enrichmentsdict
Dictionary with schema:
{ <ontology> : { [ {'rank' : <rank>, 'term' : <term>, 'pvalue' : <pval>, 'zscore': <zscore>, 'combined_score': <combined_score>, 'genes': [<gene1>, ..., <geneN>], 'adj_pvalue': <adj_pval>}, ..., ] } }
- plot_enrichments(topic_num, show_genes=True, show_top=10, barcolor='lightgrey', label_genes=[], text_color='black', plots_per_row=2, height=4, aspect=2.5, max_genes=15, pval_threshold=1e-05, color_by_adj=True, palette='Reds', gene_fontsize=10)#
Make plot of geneset enrichments results.
- Parameters
- topic_numint
Topic for which to plot results
- show_genesboolean, default = True
Whether to show gene names on enrichment barplot bars
- show_topint > 0, default = 10
Plot this many top terms for each ontology
- barcolorstr or tuple[int] (r,g,b,a) or tuple[int] (r,g,b)
Color of barplot bars
- label_geneslist[str] or np.ndarray[str]
Add an asterisc by the gene name of genes in this list. Useful for finding transcription factors or signaling factors of interest in enrichment results.
- text_colorstr or tuple[int] (r,g,b,a) or tuple[int] (r,g,b)
Color of text on plot
- plots_per_rowint > 0, default = 2
Number of onotology plots per row in figure
- heightfloat > 0, default = 4
Height of each ontology plot
- aspectfloat > 0, default = 2.5
Aspect ratio of ontology plot
- max_genesint > 0, default = 15
Maximum number of genes to plot on each term bar
- pval_thresholdfloat (0, 1), default = 1e-5
Upper bound on color map for adjusted p-value coloring of bar outlines.
- color_by_adjboolean, default = True
Whether to outline term bars with adjusted p-value
- palettestr
Color palette for adjusted p-value
- gene_fontsizefloat > 0, default = 10
Fontsize of gene names on term bars
- Returns
- axmatplotlib.pyplot.axes
Examples
>>> rna_model.post_topic(13, 500) >>> rna_model.fetch_topic_enrichments(13, ... ontologies=['WikiPathways_2019_Mouse','BioPlanet_2019']) >>> rna_model.plot_enrichments(13, height=4, show_top=6, max_genes=10, ... aspect=2.5, plots_per_row=1)
